
Developing a patient-centric and compliant study is a top priority for any clinical team. Understanding the importance of Institutional Review Boards (IRB) and Independent Ethics Committees (IEC) and their role is critical to a successful clinical trial. Before a trial can begin, however, each aspect of the study must first be reviewed and approved by a designated regulatory body. IRBs and IECs check each component of the clinical trial, from protocol development to patient reimbursement and travel support. Below, you will learn about several types of these boards and how Clincierge helps sponsors and CROs keep logistics support and reimbursement compliant to avoid any study delays.
IRBs and ECs: Frequently Asked Questions
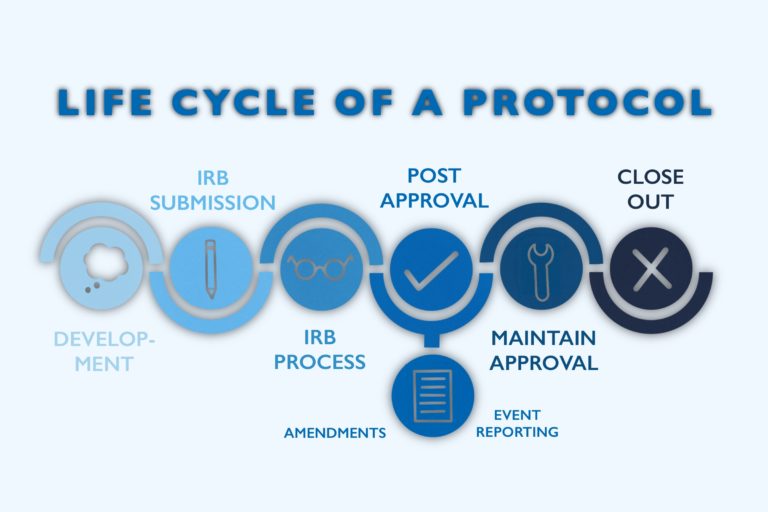
What is an Independent Ethics Committee (IEC)?
An Independent Ethics Committee comprises various pharmaceutical industry stakeholders, including doctors, nurses, social workers, chaplains, and community members. These members come from diverse backgrounds and are stakeholders in the delivery of healthcare. They join together to discuss the clinical trial’s scientific, ethical, and legal ramifications and evaluate the benefit/risk factors involved.
What is the purpose of an Independent Ethics Committee (IEC)?
Ethics Committees ensure that human subject research and medical experiments operate according to all national and international laws.
What is an Institutional Review Board (IRB)?
An Institutional Review Board is a formally designated group assigned to monitor and review any medical research involving human subjects. This team has the authority to approve or disapprove research and ask for research modifications to lead to approval. IRBs operate under United States Food and Drug Administration (FDA) regulations.
What is the purpose of an Institutional Review Board (IRB)?
Institutional Review Boards are responsible for protecting and assuring that human research subjects’ rights and welfare both before they begin their participation (with appropriate protocols in place) and throughout the clinical trial (through periodic study reviews).
What is the difference between an IRB and IEC?
Clinical trials conducted in the European Union are held accountable by Independent Ethics Committees (IECs). In the United States, Institutional Review Boards (IRBs) have oversight and must abide by the United States Food and Drug Administration (FDA) regulations. Countries other than those in the European Union and the United States have individual committees and regulations.
There are two types of IRBs and IECs, local and central. Local committees support individual research institutions and are responsible for reviewing only their clinical trials. Central boards oversee the review of clinical studies for many different organizations.
More specific guidelines on IRB and IEC responsibilities, composition, functions, and operations are listed in this addendum.
How are IRBs and IECs formed?
Per the FDA, each IRB/IEC should include five or more members and come from various backgrounds. One of these representatives should come from a non-scientific background and is referred to as a lay member. Another should be an independent member who is not affiliated with the clinical site or medical institution. All members should be competent individuals who are able to understand all aspects of the clinical trial process.
The Food and Drug Administration (FDA) has compiled a list of frequently asked questions and their corresponding answers to provide clarity on United States regulatory procedures. This document explains how IRBs are created, outlines their responsibilities, and gives detail on various questions one may encounter during the approval process.
A similar directive has been provided for countries in the European Union. Once again, the arduous clinical trial process is broken down into more manageable information, but the reader will continue to realize the numerous hurdles to be overcome.
Why are IECs and IRBs so critical?
Independent Ethics Committees and Institutional Review Boards are necessary to monitor all aspects of clinical trials, ensuring that human trial participants’ safety is their top priority. They review each study through neutral eyes, evaluating the risks and benefits throughout every clinical trial process step.
How long does it take to receive IRB/IEC approvals?
As explained earlier, each aspect of the clinical trial, including patient payment guidelines, must be approved by the IRB before any patient recruitment and enrollment are permitted to begin. Generally, an IRB submission takes one month to receive approval, with expedited/exempt approvals returned in about two weeks. If there are any details of the study that do not align with IRB standards, the IRB refuses approval and suggests amendments to receive approval. Just one minor change could result in weeks or months of delays for a clinical trial, leading to loss of money for the sponsor and loss of much-needed treatment for trial participants.
Each country across the world has its regulations for stipend and per diem payments, patient reimbursements, and tax reporting of income related to clinical trial participation. When conducting a trial at sites in various countries, understanding these unique requirements is hugely time-consuming and overwhelming. Often, countries will make changes to their guidelines, causing the need to quickly pivot to adapt to these updates and adhere to the new rules.
As your full-service provider of logistical support, Clincierge has the expertise needed to help assess the feasibility of supporting patients in specific regions worldwide. We streamline the process by providing required documents in multiple languages as well as manage any document revisions. Our team keeps abreast of current regulations in each country and provides recommendations to ensure compliance and adherence to specific guidelines.
Clincierge has the expertise needed to help you navigate the complexities involved with IRBs and IECs across the globe. These services will ease the burdens associated with your clinical trial regulatory processes. We handle:
- Information Compilation and Document Submissions
- IRB/EC Submissions
- Regulatory Reports
- Study Amendment Documentation
- Study Termination Documentation
